
THE ANNALS of EMERGENCY MEDICINE
Journal of the American College of Emergency Physicians
April 2002 • Volume 39 • Number 4
AUTHORS: Laurence M. Katz, MD*, Yuanfan Wang, MD*,
 Steve Rockoff, MSII,§, Thomas W. Bouldin, MD‡.
Steve Rockoff, MSII,§, Thomas W. Bouldin, MD‡.
CITATION:
Katz LM, Wang Y, Rockoff S, Bouldin TW. Low-dose Carbicarb improves cerebral outcome after asphyxial cardiac arrest in rats. Ann Emerg Med (United States), Apr 2002;39(4): p359-365.

Abstract:
Study Objective: Controversy surrounds the use of buffers during cardiac arrest to correct acidosis. The objective of this study was to determine whether attenuation or neutralization of cerebral acidosis by Carbicarb alters hippocampal glutamate levels, neuronal cell death, and neurologic deficits after reperfusion from asphyxial cardiac arrest in rats.
Methods: Rats were prospectively randomized to either a control (n=45), low-dose Carbicarb (LDC; 3 mL/kg, n=45), or high-dose Carbicarb (HDC; 6 mL/kg, n=45) group in a blinded fashion during resuscitation after 8 minutes of asphyxial cardiac arrest. Microdialysis was used to assess brain pH and glutamate. A neurologic deficit score and neuronal cell death in the hippocampus were determined at day 7.
Results: Resuscitation was greatest in LDC rats (42/45) and least in HDC rats (28/45) versus that in control rats (34/45). Brain pH was higher in the LDC and HDC rats 10 minutes after resuscitation and remained higher than that of control rats for 120 minutes after resuscitation. Glutamate levels at 10 to 120 minutes after reperfusion were lowest in the LDC rats. LDC rats had the lowest neurologic deficit score (1±2) versus that of control rats (13±8) and HDC rats (19±6). Hippocampal neuronal cell death was lowest in LDC rats (30±20) versus that in control rats (86±47) and HDC rats (233±85).
Conclusion: LDC administered during resuscitation from asphyxial cardiac arrest attenuated acidosis, improved resuscitation, and reduced neurologic deficits and the number of dead hippocampal neurons. Neutralization of cerebral acidosis with HDC increased the number of dead hippocampal neurons and neurologic deficits after resuscitation from cardiac arrest in rats.
Cerebral ischemia leads to tissue acidosis during cardiac arrest, and reperfusion and has been correlated with neuronal death.(1) Oxidative stress caused by free radicals(2) and the release of excitatory amino acids (EAAs)(3) have been implicated as pathophysiologic mechanisms of selective neuronal death during reperfusion from global ischemia. Ischemia-induced acidosis increases oxidative stress in the brain by increasing the availability of iron necessary for production of free radicals by means of the Fenton reaction.(4) In vitro, acidosis has been variously reported to increase the toxicity of EAAs by decreasing glutamate reuptake (5) or to provide neuroprotection from the toxic effects of glutamate.(6) In vivo, the effects of buffering cerebral acidosis on glutamate levels and neurotoxicity are even less clear. Because cerebral acidosis influences the activity of both free radicals and EAAs, it may be an important upstream mechanism involved with modulation of glutamate and neuronal cell death.
Asphyxia is the most common cause of cardiac arrest in children and a frequent cause in adults.(7) Asphyxia leads to retention of carbon dioxide, hypoxic ischemia, and cerebral acidosis. These physiologic changes may contribute to worse cerebral outcome compared with that of ventricular fibrillation of the same duration.(8) Therefore, in addition to reversal of asphyxia, supplemental therapeutic interventions may be needed to reduce brain damage from asphyxial cardiac arrest.
Use of the buffer sodium bicarbonate during cardiac arrest has been discouraged because it generates carbon dioxide and produces a paradoxical cerebral acidosis.(9) However, reevaluation of the use of a buffer during reperfusion from cardiac arrest has shown a beneficial effect on cardiovascular function that can positively influence cerebral outcome.(10) Presumably, the positive effect on cerebral perfusion outweighs the risk of worsening cerebral acidosis. Carbicarb is a formulation of sodium bicarbonate and sodium carbonate designed to neutralize systemic and tissue acidosis without producing carbon dioxide.(11) Carbicarb effectively neutralizes cerebral acidosis during cardiac arrest without producing a paradoxical acidosis, but its effects on cerebral outcome are less certain.(12)
The purpose of this study was to determine whether attenuation or neutralization of cerebral acidosis by Carbicarb alters hippocampal glutamate levels, neurologic deficits, and neuronal cell death after reperfusion from asphyxial cardiac arrest in rats. We hypothesized that low-dose Carbicarb (LDC) improves neurologic outcome. We also hypothesized that high-dose Carbicarb (HDC) increases neurologic deficits and neuronal cell death in survivors.
Materials and Methods
The Institutional Animal Care and Use Committee of the University of North Carolina School of Medicine approved the protocol. The study was prospectively randomized in a blinded fashion and was conducted in a university research laboratory. One hundred thirty-five rats were randomized to a control (n=45), LDC (3 mL/kg, n=45), or HDC (6 mL/kg, n=45) group. The control group was further divided into 2 groups that received equal volumes and concentrations of sodium chloride (NaCl) that paralleled the LDC (3 mL/kg of 1 mmol/mL, n=22) or HDC (6 mL/kg of 1 mmol/mL, n=23) groups. Rats were prepared for cardiac arrest and reperfusion, as previously described.(13) Briefly, rats were anesthetized with 4% isoflurane, intubated, and mechanically ventilated with a combination of 30% oxygen and 70% nitrous oxide. Titrated isoflurane anesthesia was maintained throughout the preparation phase. Catheters were placed in the left femoral vessels for monitoring of mean arterial pressure (MAP), withdrawal of blood for arterial blood gas measurement, and administration of intravenous medications. A telemetric temperature probe (Mini Mitter, Bend, OR) was stereotactically placed in the epidural space, and brain temperature was monitored and maintained at 37.2°C (99°F) throughout the experiment. A microdialysis catheter was placed in the left hippocampus for sampling of extracellular pH and glutamate during baseline and asphyxia and for 120 minutes of reperfusion. Rats were chemically paralyzed with vecuronium (1 mg/kg administered IV), and apneic asphyxia was induced by discontinuation of ventilation. Asphyxia led to cardiac arrest within approximately 4 minutes in all rats, and asphyxia was maintained for 8 minutes. Rats were resuscitated with epinephrine (0.005 mg/kg administered IV), study drug (1 mL/kg), mechanical ventilation with 100% oxygen, and chest compressions. Chest compressions were stopped when there was a return of spontaneous circulation (ROSC) (MAP  60 mm Hg) or no ROSC after 2 minutes. Attempts at resuscitation were discontinued after 2 minutes because in a prior study, rats that had resuscitation for longer than 2 minutes rarely survived the 72 hours required for neurologic testing.(13) An infusion of study drug was continued for 2 hours after ROSC. Control rats received a 1 or 2 mL/kg bolus of NaCl (1 mmol/mL) during resuscitation and 2 or 4 mL/kg of NaCl (1 mmol/mL) over 2 hours during reperfusion.
60 mm Hg) or no ROSC after 2 minutes. Attempts at resuscitation were discontinued after 2 minutes because in a prior study, rats that had resuscitation for longer than 2 minutes rarely survived the 72 hours required for neurologic testing.(13) An infusion of study drug was continued for 2 hours after ROSC. Control rats received a 1 or 2 mL/kg bolus of NaCl (1 mmol/mL) during resuscitation and 2 or 4 mL/kg of NaCl (1 mmol/mL) over 2 hours during reperfusion.
Syringes were filled with study drug in a random manner, as determined by using the HP-11c (Hewlett-Packard, Palo Alto, CA) computer random-number generator. A person blinded to the study protocol administered the study drug. The HP-11c computer random-number generator was also used to determine which study drug would be administered. The LDC group received a 1 mL/kg bolus (NaCl concentration, 1 mmol/mL) during resuscitation and 2 mL/kg over 2 hours during reperfusion. The HDC group received a 2 mL/kg bolus during resuscitation and 4 mL/kg during reperfusion. A person blinded to the study protocol administered the study drug. The investigator (YW) who performed animal surgery, cardiac arrest, and resuscitation was blinded to the contents or volume of the study drug, as well as the results of blood gases and microdialysis values. Rats were extubated 2 hours after ROSC and returned to a temperature-controlled environment, in which the brain temperature was maintained at 37.2°C (99°F). Rats had free access to food and water during recovery.
A microdialysis probe (CMA/10; membrane length 4 mm; diameter 0.5 mm; Carnegie Medecin, Stockholm, Sweden) was placed in the left hippocampus under stereotactic guidance (Bregma 3.4 mm caudal and 2 mm lateral to the sagittal sinus and 3 mm ventral to the skull). Artificial cerebrospinal fluid (Na+, 145 mmol/L; Ca2+, 2.75 mmol/L; Cl-, 137.5 mmol/L; HCO3, 1 mmol/L; pH 7.0) was perfused at 2 µL/min, and 20-µL samples were collected at 30, 20, and 10 minutes before asphyxia; during asphyxia (collected 1 minute before asphyxia and continued for 1 minute after ROSC); and 10, 20, 30, 40, 50, 60, 90, and 120 minutes after reperfusion in rats in which ROSC was achieved. The dialyzed samples were measured in line for pH, as described by Landolt et al.(14) Dialysis samples were then immediately sampled for glutamate levels by means of high-pressure liquid chromatography.
Glutamate was measured with a high-pressure liquid chromatography system (Applied BioSystems 130A Separation System; Applied Biosystems, Foster City, CA) with fluorescence detection and precolumn derivatization. The precolumn derivatization was performed with phenylisothiocyanate reagent, and the injections were performed automatically with an autoinjector (Applied BioSystems 420A Derivatizer; Applied Biosystems). The elution of glutamate was achieved with a phosphate mobile phase (0.15 mol/L phosphate, pH 6.8, adjusted with 1N NaOH containing 30% methanol and 25 mg/L ethylenediamine tetraacetic acid) at a flow rate of 1 mL/min.
A neurologic deficit score (NDS) was obtained daily for 7 days after ROSC by an investigator (LMK) blinded to the treatment. Rats were tested for coordination (balance beam walk, placing test, depth perception, and righting reflex) and for motor and sensory function, as previously described.(13) The NDS ranges from 0 (normal) to 100 (brain dead).
Seven days after ROSC, the rats were reanesthetized with 4% isoflurane and mechanically ventilated. A needle was placed in the left ventricle and advanced into the ascending aorta proximal to the carotid arteries. The descending aorta was crossclamped, and 4% buffered paraformaldehyde was infused under a pressure of 100 cm of water, for a total of 100 mL. The rats were decapitated, and the heads were placed in paraformaldehyde at 4°C for 24 hours before the brains were removed. The brains were embedded in paraffin, and 6-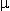 m-thick ... 6-µm-thick sections were cut, stained with hematoxylin-eosin, and examined by means of light microscopy. The pathologist (TWB) was blinded to the treatment group. The total number of neurons showing ischemic cell death in one standardized section of the left and right cornu ammonis of the hippocampus (CA1-CA4 region) was counted. Neurons were considered to show acute ischemic cell death if the cytoplasm was eosinophilic and the nucleus was pyknotic or karyorrhectic.(15)
m-thick ... 6-µm-thick sections were cut, stained with hematoxylin-eosin, and examined by means of light microscopy. The pathologist (TWB) was blinded to the treatment group. The total number of neurons showing ischemic cell death in one standardized section of the left and right cornu ammonis of the hippocampus (CA1-CA4 region) was counted. Neurons were considered to show acute ischemic cell death if the cytoplasm was eosinophilic and the nucleus was pyknotic or karyorrhectic.(15)
A power analysis determined that 27 animals in each group would be required to detect a 25% difference in neurologic outcome between groups (the primary end point of the study) with a power of 0.8. A 25% difference in neurologic outcome was believed to have some clinical relevance. Forty-five rats were entered into each group, with anticipation that ROSC/survival would be approximately 60%. Baseline physiologic variables, brain pH, glutamate levels, and the number of dead neurons were reported as means±SDs, and the number of dead neurons were analyzed by using 2-way analysis of variance. Post hoc analysis was performed by means of Tukey analysis if there was a significant difference between groups. Kruskal-Wallis analysis was used to assess NDS. ROSC was analyzed by using the X2 test. Significance was set at a P value of less than .05. Data were entered and analyzed with SPSS software (SPSS, Chicago, IL).
Results
The baseline brain temperature (37.2°±0.2°C [99°±0.4°F]), MAP (110±8 mm Hg), blood glucose (121±10 mg/dL), arterial blood gases, and extracellular brain pH (7.00±0.04) were similar among groups. There were no differences in outcome parameters between control rats that received 3 and 6 mL/kg volumes during resuscitation, and therefore, their values were combined for comparisons with the LDC and HDC groups. There were no group differences in time to cardiac arrest (252±44 seconds; 95% confidence interval [CI] 242 to 262) or ROSC (34±8 seconds; 95% CI 27 to 38). Thirty-four of 45 control rats and 28 of 45 HDC rats had ROSC; a significantly larger number of LDC rats (42/45) had ROSC compared with the control and HDC group (P<.05, X2 test).
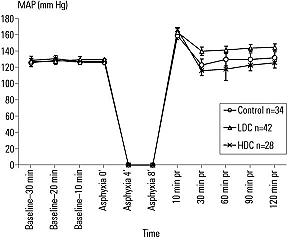
All rats that had ROSC survived 7 days and were analyzed for reperfusion data. Administration of LDC or HDC did not increase arterial PCO2 during reperfusion (data not shown). MAP at 10 to 120 minutes after ROSC was higher in the LDC group than in the control or HDC groups. (Figure 1)
The HDC group had the highest arterial blood pH during reperfusion compared with that seen in the control and LDC groups. (Figure 2)
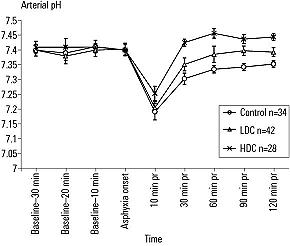
Brain pH in the LDC and HDC groups was higher than that in control rats 10 to 120 minutes after ROSC. (Figure 3)
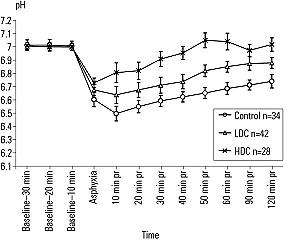
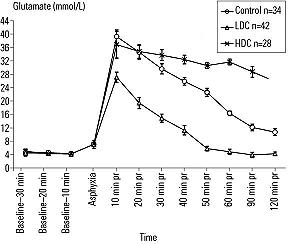
Brain pH in the LDC group remained lower than that in the HDC group but higher than that in the control group at all time points after ROSC. Glutamate levels 10 to 120 minutes after reperfusion were lower in the LDC group compared with those in the control or HDC groups and higher in the HDC group compared with those in the control or LDC groups 30 to 120 minutes after reperfusion. (Figure 4)
NDSs were lowest in the LDC group (1±2) and highest in the HDC group (19±6) when compared with those in the control group (13±8) at 7 days after ROSC (P<.05; Kruskal-Wallis test). Neurons showing ischemic cell death were fewest in the LDC group (30±20; 95% CI 11 to 45) and most numerous in the HDC group (233±85; 95% CI 150 to 298) when compared with those in the control group (86±47; 95% CI 57 to 119; P<.05, analysis of variance and post hoc Tukey test).
Discussion
Therapeutic interventions during reperfusion from hypoxia-ischemia have the potential to influence cerebral outcome because a significant proportion of damage occurs during reperfusion.(16) The difficulty in developing therapies is that multiple metabolic derangements influence cerebral outcome, and thus, treatment of one abnormality may exacerbate another.(17) A second consideration during development of a cerebral therapy is its influence on the cardiovascular system. Therapies that improve cerebral outcome in the laboratory have failed to improve cerebral outcome in human patients, most likely because of the therapy's negative cardiovascular side effects.(18) Buffer therapy was studied because pH modulates multiple metabolic and cardiovascular processes and thus has the potential for beneficial effects on multiple organ systems.
In 1974, the buffer sodium bicarbonate was recommended for administration during cardiac arrest because acidosis was believed to be detrimental to outcome.(19) Concern about the use of a buffer during resuscitation from cardiac arrest arose from laboratory research demonstrating that administration of sodium bicarbonate without epinephrine during cardiac arrest caused increased production of carbon dioxide in the blood, which then diffused to the heart and brain and caused a paradoxical acidosis.(20) The results from these experimental models were extrapolated to clinical practice, and sodium bicarbonate administration was discouraged during cardiac arrest in 1986.(21) However, recent studies indicate that sodium bicarbonate, in combination with epinephrine, improves neurologic outcome in a survival model of cardiac arrest in rats and dogs.(22, 23) The results of our study, which used a different buffer that does not cause a paradoxical acidosis, are in agreement with these recent studies and further demonstrate that the dosage of buffer is critically important in determining the effect on neurologic outcome.
Severe acidosis has been associated with poor outcome after cerebral ischemia.(1) Acidosis has a direct toxic effect on neuronal metabolism, cell membrane integrity, and recovery of energy metabolism necessary for cell viability after ischemia.(24) However, it has been hypothesized that less severe acidosis may be beneficial to neuronal viability during ischemia and reperfusion by providing resistance to damage mediated by glutamate.(6) The results of our study support this hypothesis. The LDC group was still acidotic, but less so than the control group, and had the least ischemic damage in the hippocampus and lowest NDSs of all groups. In contrast, the HDC group, which had complete neutralization of the systemic and brain acidosis, had the most ischemic neuronal damage and highest NDSs of all groups.
Altering brain pH with Carbicarb during reperfusion affected glutamate levels. The study design does not allow determination of whether LDC affected glutamate levels by reducing release or increasing reuptake of glutamate. Both mechanisms are responsible for the total level of glutamate during reperfusion from ischemia, and it is possible that the buffer affected both pathways.(25) HDC neutralized hippocampal pH and was associated with increased glutamate levels and increased ischemic neuronal cell death. This demonstrates that the amount of buffer, and thus the severity of cerebral acidosis, is associated with brain glutamate levels after reperfusion from cardiac arrest.
LDC had a beneficial effect on blood pressure during reperfusion from asphyxial cardiac arrest. These results are consistent with those reported by Sun et al.(26) Improved blood flow during reperfusion has been associated with improved neurologic outcome after resuscitation from cardiac arrest.(27) The cardiovascular effect of Carbicarb may have contributed to the beneficial neurologic outcome, but it is unlikely to be the sole variable because the rats were not hypertensive during reperfusion, as required in the flow promotion study.(27) In addition, capillary hypoperfusion after cerebral ischemia is unlikely to be improved by increasing MAP during reperfusion because of the loss of autoregulation early after cerebral ischemia. Furthermore, the HDC group had a worse outcome, even though the blood pressures in this group were similar to those in the control group.
The dose of Carbicarb affected the ability to achieve ROSC. This result is not unexpected because large loads of sodium decrease coronary perfusion pressure by decreasing diastolic pressure and lowering the rate of ROSC.(10) In contrast, the improved rate of ROSC in the LDC rats may reflect a synergistic effect of epinephrine and buffer on cardiac function.(28)
LDC given to rats during reperfusion from asphyxial cardiac arrest improved neurologic outcome, as demonstrated by reduced histologic damage in the hippocampus and the lowest (best) NDSs. In contrast, HDC caused the worse neurologic outcome. The NDS, a crude behavioral evaluation of overall brain function, has been correlated with global histologic brain damage.(13) The NDS combined evaluation of cranial nerves, limb function, and performance on coordination tests. Scores improved consistently from days 1 to 3 and remain unchanged from days 3 to 7 (data not shown). All animals received the same handling and analysis after reperfusion to minimize potential bias of conditioning or learning. The HDC rats were awake, ambulatory, and able to eat and drink but were unable to walk on a balance beam, were unable to right themselves, failed a placing test, and had no depth perception. In contrast, the performance of the LDC rats was nearly normal.
A limitation of the study was that extracellular brain pH and glutamate levels were measured rather than intracellular levels. This was necessary because microdialysis was used for sampling. However, it has been shown that during cardiac arrest and early reperfusion, intracellular and extracellular pH equilibrate between compartments because of energy failure.(29) The microdialysis method also allowed for determination of the temporal relationship between pH manipulation and glutamate levels. An additional limitation was that only 1 region of the brain was analyzed in this model of global brain ischemia, and it is likely that other regions would have differing amounts of damage. However, the hippocampus in this and most other cardiac arrest models has been shown to be the region most vulnerable to ischemia.(30) Moreover, the changes observed in the hippocampus correlated with changes that were observed in the NDS, a more global assessment of neurologic function. Finally, asphyxia in this model of cardiac arrest does not cause sudden cessation of cardiac function, as in other animal models of cardiac arrest with a more precise onset of the insult. Despite the inexact nature of asphyxial cardiac arrest, the duration of low flow (cardiac arrest and CPR time) and, most importantly, cerebral outcome was reproducible in this model.
In conclusion, LDC administered during resuscitation from asphyxial cardiac arrest attenuated cerebral acidosis, improved resuscitation, and reduced neurologic deficits and the number of dead hippocampal neurons. Paradoxically, neutralization of cerebral acidosis with HDC increased neurologic deficits and the number of dead neurons in the hippocampus after resuscitation from asphyxial cardiac arrest in rats.
We thank Leisa Runquist for her editorial review of the manuscript.
References:
Publishing and Reprint Information
 Email: LKatz@med.unc.edu
Email: LKatz@med.unc.edu

Email Charly at: c-d-miller@neb.rr.com